Biology DivisionLaboratory of Microbial Carcinogenesis
INDEX
- Deregulation of the prooncogenic phosphatase SHP2 and inhibition of the cell polarity regulator PAR1b by CagA
- CagA polymorphism and gastric cancer
- Oncogenic activity of CagA in vivo
- 3D structure of CagA protein
- Reprogramming and dedifferentiation of gastric cells by CagA
- SHP1 as a suppressor for CagA oncogenic acticvity: Cooperation of bacteria with virus in gastric cancer
- Hit-and-run gastric carcinogenesis mediated by the CagA-driven "extrinsic BRCAness"
- Parafibromin, a novel substrate of SHP2
- Identification of an intracellular "logic board" implemented by Parafibromin
- Subcellular localization and functions of SHP2 controlled by cell density
- Collapse of Hippo pathway via hyaluronan degradation
- Distinct pro-oncogenic potentials of YAP1 regulated by alternative splicing
Research Review
1. Deregulation of the prooncogenic phosphatase SHP2 and inhibition of the cell polarity regulator PAR1b by CagA
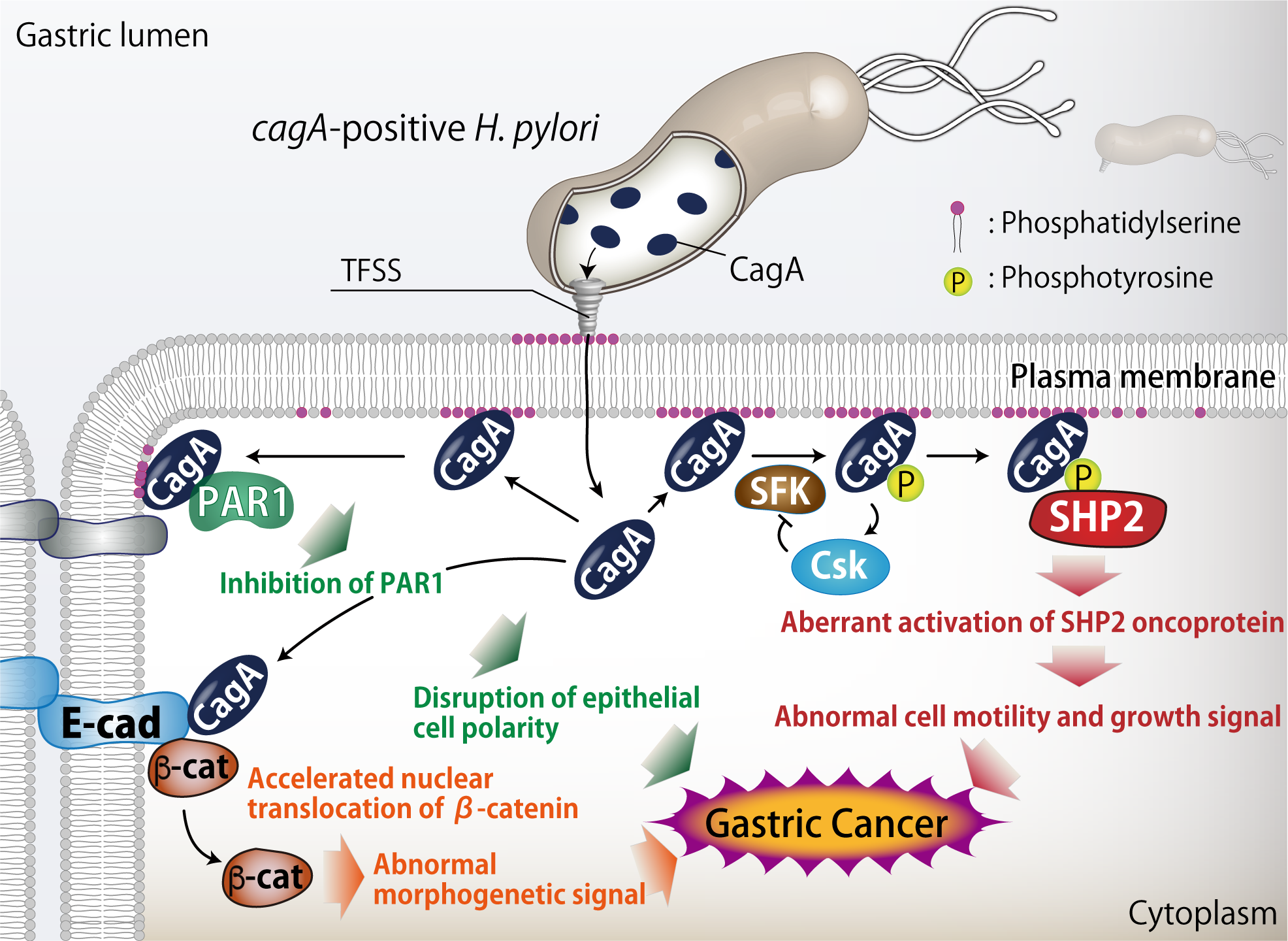
After attachment to gastric epithelial cells, H. pylori cagA-positive strains inject the CagA protein directly into the cells via the type IV secretion system. Injected CagA then undergoes tyrosine phosphorylation by Src family kinases. We found that tyrosine phosphorylated CagA binds to Src homology 2-containing protein tyrosine phosphatase 2 (SHP2), a bona-fide human oncoprotein. We also found that the CagA-SHP2 complex inactivates focal adhesion kinase (FAK), resulting in the morphological transformation known as the hummingbird phenotype. In addition, CagA-activated SHP2 deregulates the RAS-MAP kinase cascade to elicit abnormal mitogenic signal.
In the stomach, gastric epithelial cells form a polarized epithelial monolayer with highly developed tight junctions. Expression of CagA in polarized epithelial cells causes junctional and polarity defects. We found that this CagA activity is mediated by the specific interaction of CagA with partitioning-defective 1b (PAR1b) serine/threonine kinase, which acts as a master regulator in establishment and maintenance of cell polarity. We showed that association of CagA inhibits PAR1b kinase activity while preventing atypical PKC-mediated PAR1b phosphorylation, collectively causing junctional and polarity defects. We also demonstrated that CagA binds to PAR1b via the 16-amino-acid sequence, which we identified as the CagA-multimerization sequence (CM sequence). We found that PAR1b-mediated CagA dimerization is important for the subsequent complex formation between CagA and SHP2. Our findings indicate that the CagA-PAR1b interaction is not only crucial in the disorganization of epithelial layer but also critically involved in the tyrosine phosphorylation-dependent CagA-SHP2 interaction. The finding uncovered a here-to-fore unrecognized link between the PAR polarity regulating system and the human pathogen H. pylori.
We also discovered that CagA directly interacts with membrane phosphatidylserine, which is present at inner leaflet of plasma membrane and exposed to outside of cells at the site of H. pylori attachment, to intrude into host cells. Inside the cells, CagA again exploits phosphatidylserine to localize to inner surface of plasma membrane, where it exhibits the oncogenic activity.
Related papers: Higashi et al., Science (2002) / Higashi et al., PNAS (2002) / Tsutsumi et al., JBC (2003) / Higashi et al., JBC (2004) / Hatakeyama, Nat Rev Cancer (2004) / Tsutsumi et al., Mol Cell Biol (2006) / Naito et al., Gastroenterology (2006) / Murata-Kamiya et al., Oncogene (2007) / Saadat et al., Nature (2007) / Murata-Kamiya et al., Cell Host Microbe (2010), etc...
2. CagA polymorphism and gastric cancer
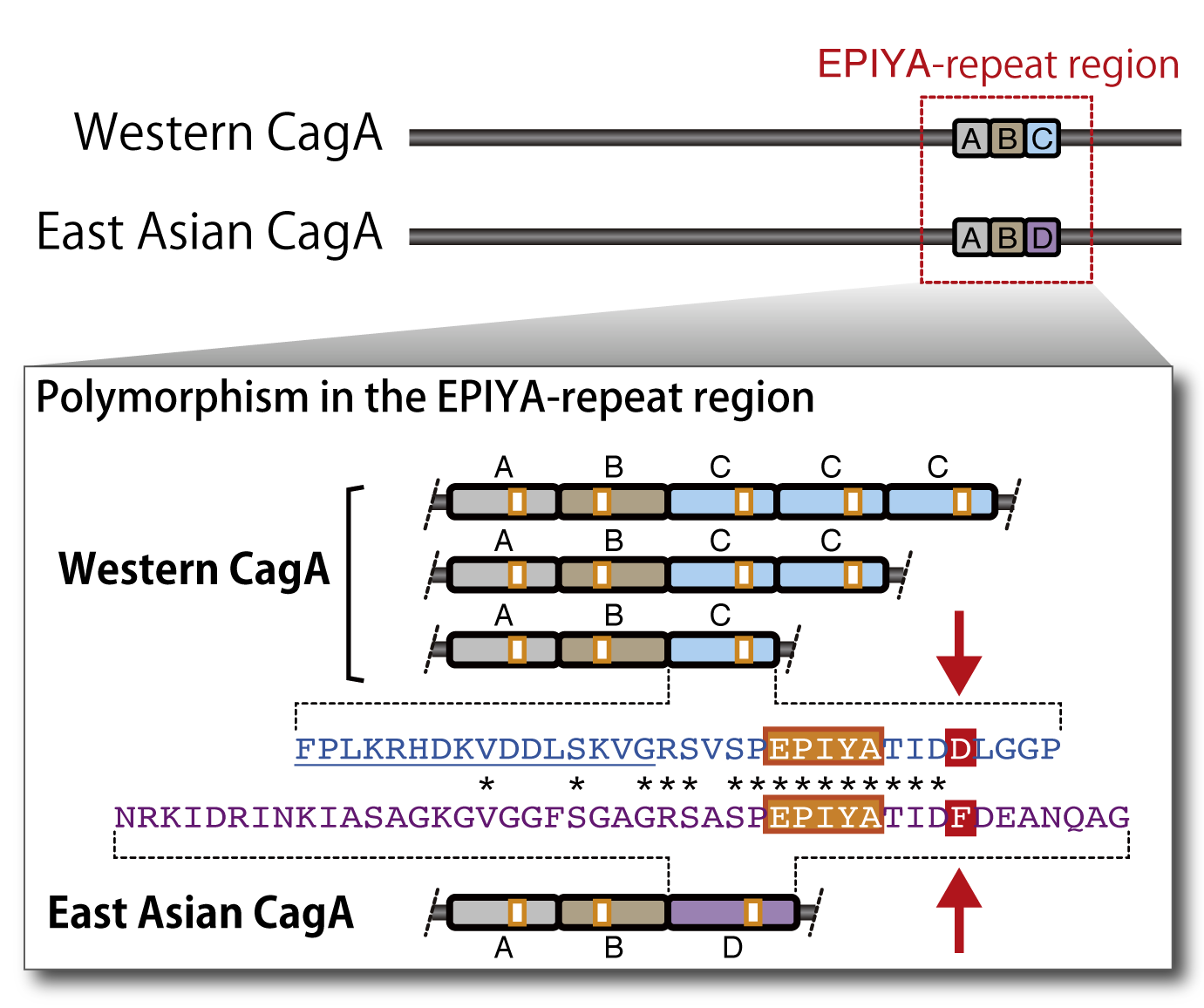
Gastric carcinoma is the second-most common cause of malignancy-related deaths worldwide and responsible for 10.4% mortality of all fatal cancer cases. The mortality rates of gastric carcinoma in East Asian countries including Japan are several times higher than those in Western countries. The H. pylori genome is highly dynamic, generating a huge diversity of strains, with each strain showing significant differences in their genome sequence. We found that the cagA gene exhibits genetic polymorphism between those derived from Western and East Asian H. pylori strains. Intriguingly, East Asian CagA exhibits stronger SHP2 binding activity and greater morphogenetic activity than Western CagA. These observations indicate that CagA-deregulated SHP2 plays a key role in the development of gastric carcinoma and that the cagA polymorphism is considered to be associated with the geographical variations in the incidence of gastric carcinoma.
Related papers: Higashi et al., Proc Natl Acad Sci, USA (2002) / Naito et al., Gastroenterology (2006)
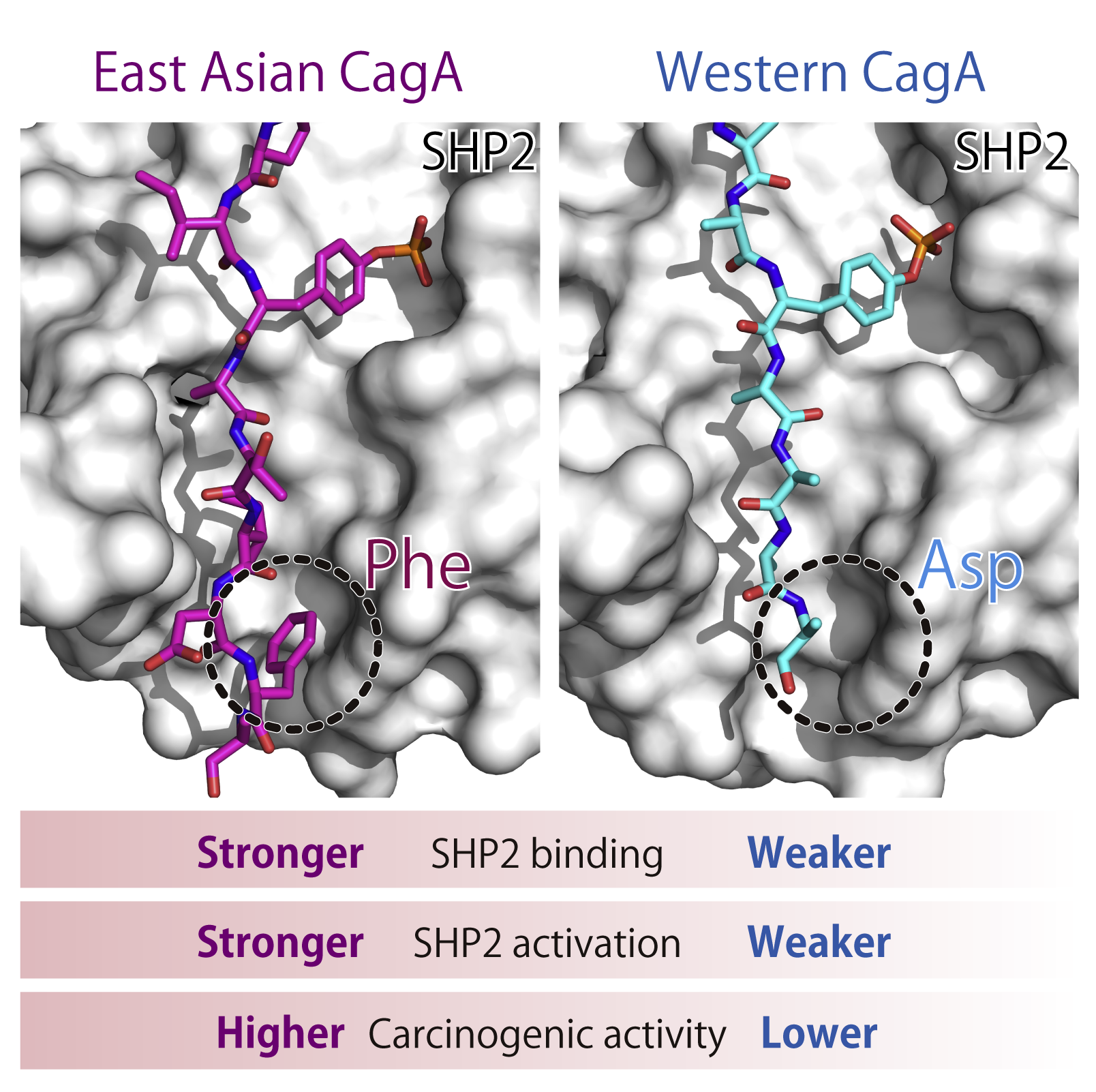
To discover the structural basis underlying the distinct oncogenic activities between East Asian CagA and Western CagA, we investigated the 3D structure of CagA-SHP2 binding interface. X-ray crystallography revealed a key structural difference due to single amino acid polymorphism in CagA. Combined with biochemical and biological data, we demonstrated that Phe at the pY+5 position in East Asian CagA acts as a barb that stabilizes the complex, whereas barbless Western CagA that possesses Asp at the pY+5 position easily dissociates from SHP2.
Related paper: Hayashi et al., Cell Reports (2017)
3. Oncogenic activity of CagA in vivo
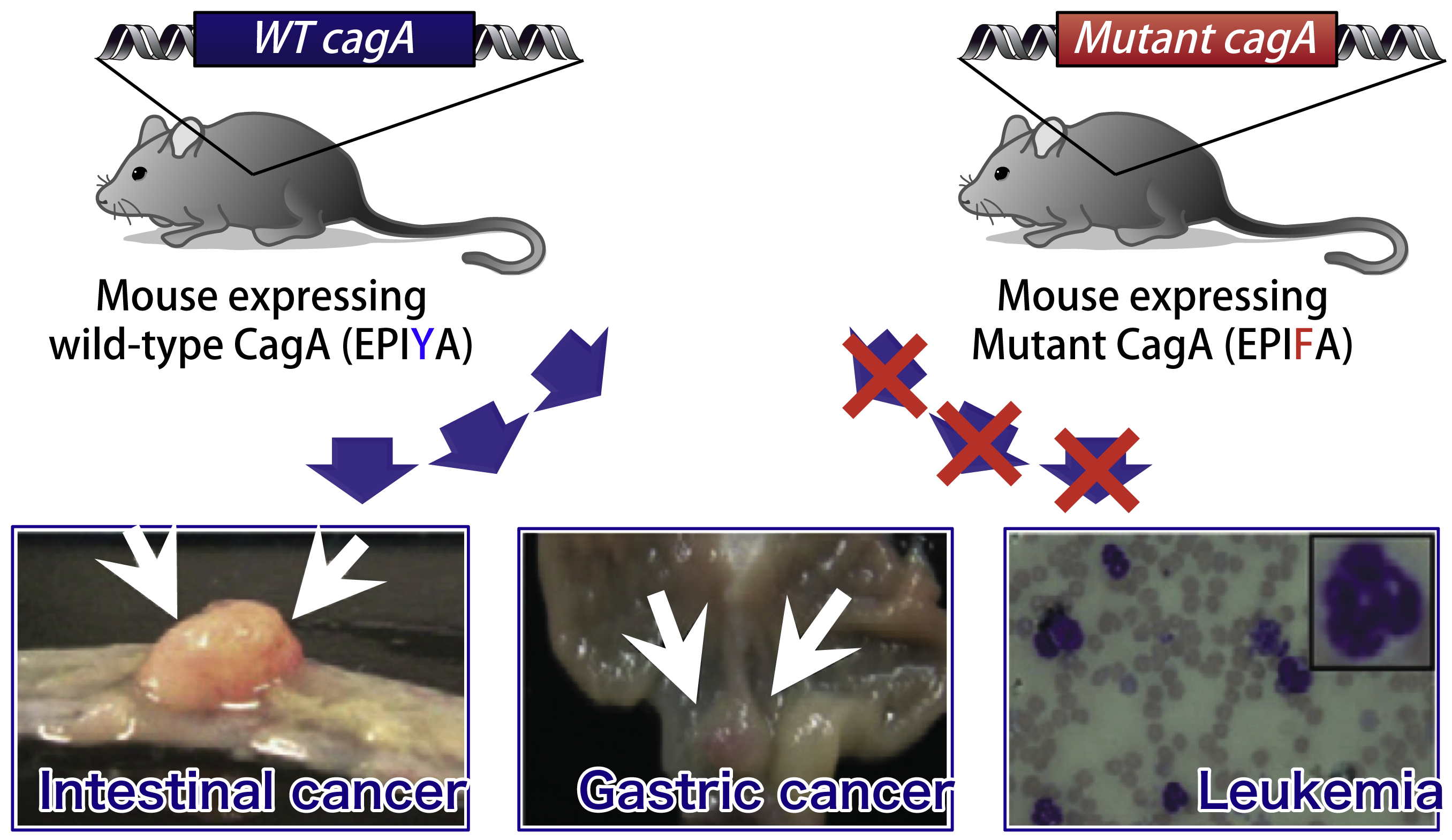
To investigate the role of CagA in in vivo carcinogenesis, we generated transgenic mice that systemically express CagA (cagA-Tg mice). Established cagA-Tg mice showed gastric epithelial hyperplasia and granulocytosis in the peripheral blood, which were followed by the development of gastric carcinoma, intestinal carcinoma and hematological malignancies such as myeloid leukemias and B cell lymphomas. Intriguingly, transgenic mice expressing phosphorylation-resistant CagA did not show any pathological abnormalities, indicating CagA-deregulated SHP2 plays an important role in the development of H. pylori-associated neoplasms. These results provide formal proof for the oncogenic potential of CagA activity as the first bacterial oncoprotein.
Related papers: Ohnishi et al., Proc Natl Acad Sci, USA (2008) / Miura et al., Int J Cancer (2009)
4. 3D structure of the CagA protein
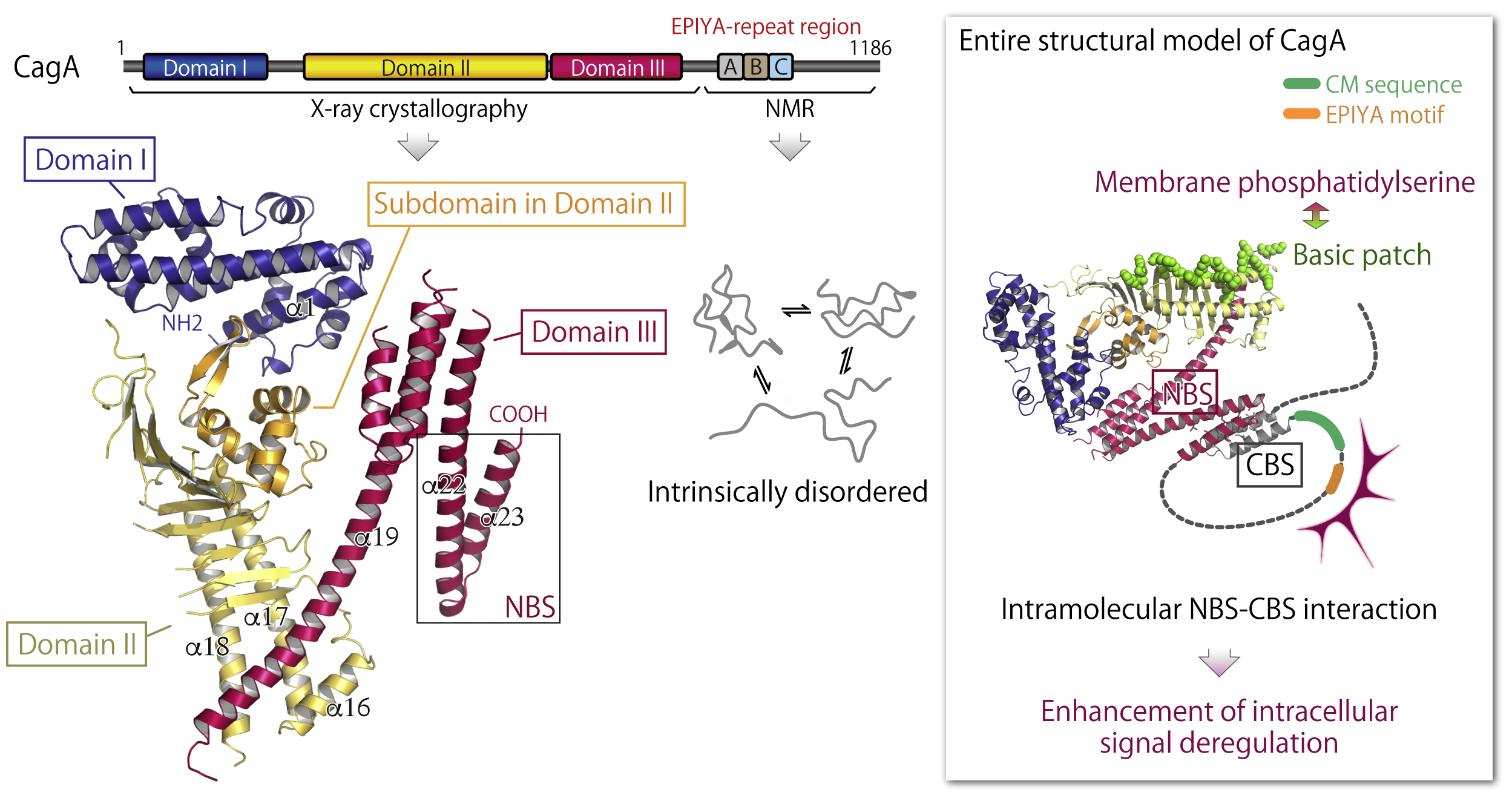
Because of a technical difficulty of the structural analysis for CagA protein, which has no homology with any of known proteins, the 3D structure of CagA remained unclear. As a result of our intensive analysis, we succeeded in elucidating the unprecedentd structure of CagA N-terminal region comprising three discrete domains (Domain I-III) by X-ray crystallography. In addition, NMR analysis revealed that the CagA C-terminal portion containing the EPIYA-repeat region shows "intrinsically-disordered structure". Since intrinsically-disordered region can flexibly alter its structure to match with interaction partners, such a frexible feature is probably important for CagA to promiscuously interact with multiple host proteins.
We also found the "basic patch" on the Dmain II that is composed of cluster of multiple basic amino-acids (Lys and Arg) and plays an important role for interaction with membrane phosphatidylserine. Furtheremore, we discovered the laliat-like loop structure formation of CagA in which "NBS" in the Domain III intramolecularly interacts with "CBS" in the C-terminal region. By forming the laliat-like structure, CagA efficiently interacts with SHP2 and PAR1b to evoke intracellular signal perturbation.
Related paper: Hayashi et al., Cell Host Microbe (2012)
5. Reprogramming and dedifferentiation of gastric cells by CagA
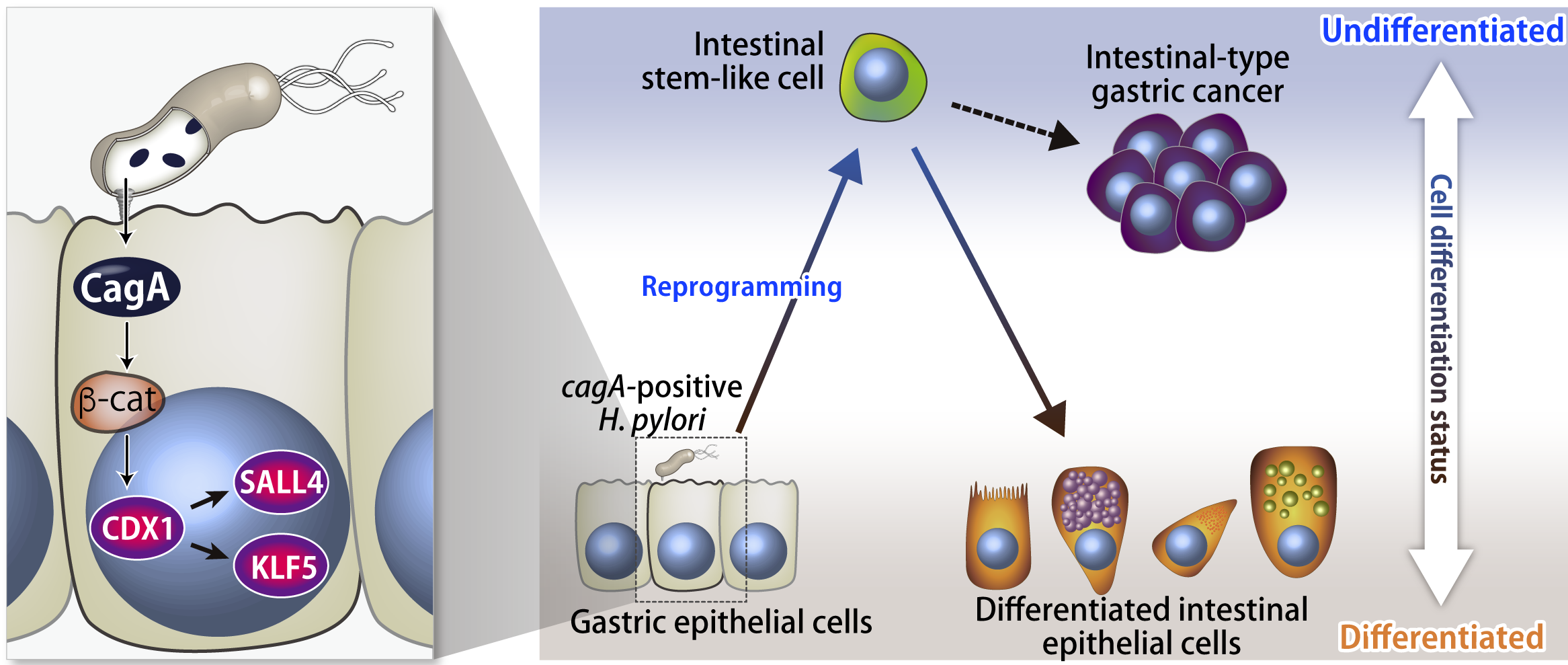
We found that CagA protein delivered into cells induces expression of a transcription factor CDX1 via promoting nuclear localization of β-catenin. Further analysis revealed that the induced CDX1 directly activates the transcription of reprogramming factors, SALL4 and KLF5 genes.
Whereas intestinal-type gastric carcinoma is a typical subtype of stomach cancer, the molecular mechanism how gastric cells acquire the intestinal cell features remained to be elucidated. In general, cells maiking up a body of multicellular organism unidirectionally differentiate from less differentiated cells to differentiated cells and normal cells don't show a retrodirective "dedifferentiation". However, we discovered that SALL4 and KLF5 induced by CagA can reprogram the cell differentiation status thereby cause retrodirective alteration of cell fate from well-differentiated gastric epithelial cells to less-differentiated intestinal stem-like cells. This finding is a first demonstration of the molecular mechanism underlying the intestinalization of gastric cells that is closely associated with intestinal-type gastric carcinogenesis.
Related paper: Fujii et al., Proc Natl Acad Sci, USA (2012)
6. SHP1 as a suppressor for CagA oncogenic acticvity: Cooperation of bacteria with virus in gastric cancer
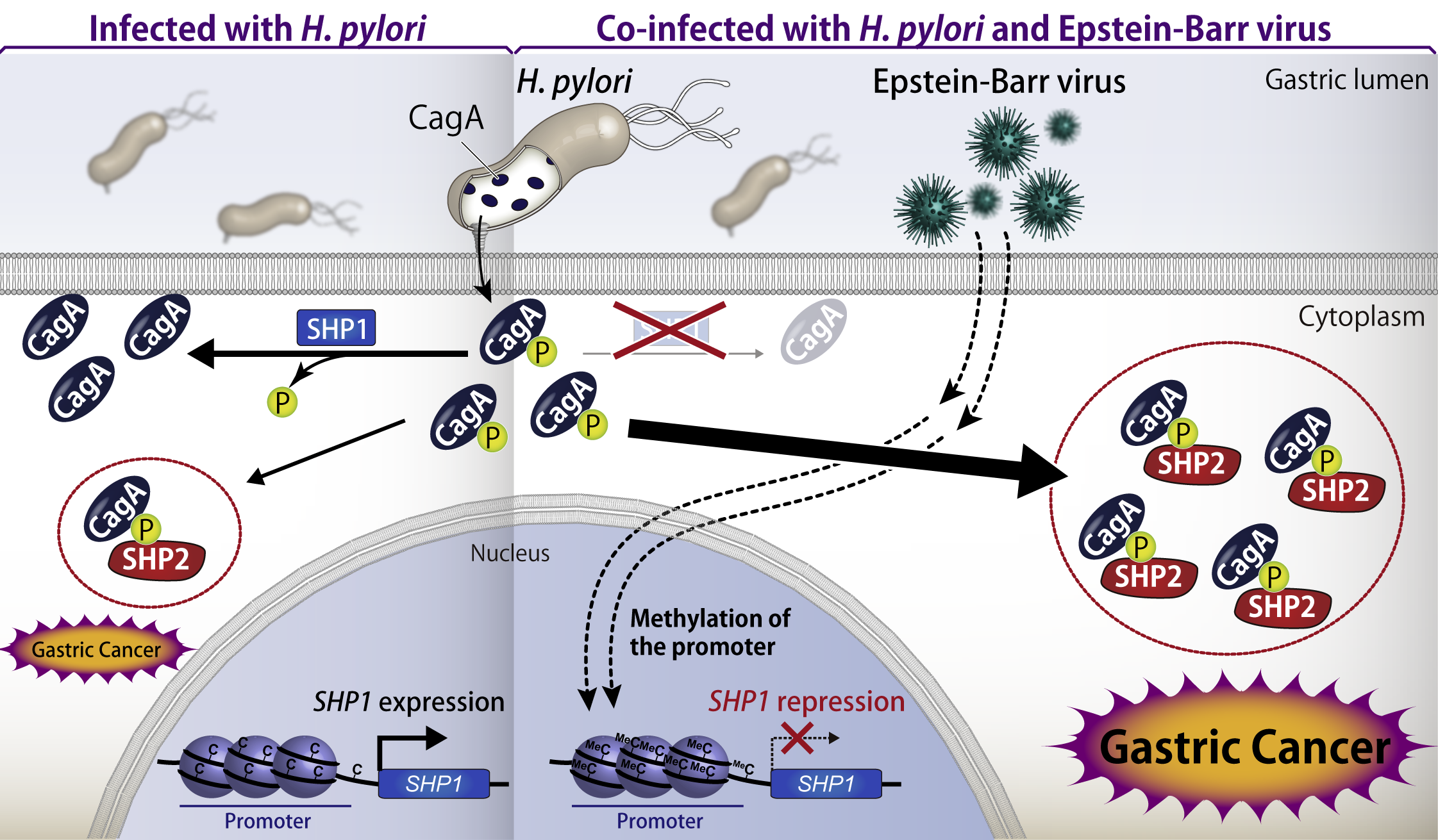
As mentioned above, CagA binds to and deregulates SHP2 in a tyrosine phosphorylation-dependent manner. We identified tyrosine phosphatase SHP1, an SHP2 paralogue, as a CagA suppressor that inhibits CagA-SHP2 interaction by specifically dephosphorylating CagA. Since the CagA-SHP2 interaction plays a crucial role in gastric carcinogenesis, specific activation of SHP1 may control the gastric cancer development.
All the Japanese cases of gastric carcinoma are considered as H. pylori-related diseases. In addition, Epstein-Barr virus (EBV) has been also suspected to be associated with gastric cancer. It is reported that 10% of H. pylori-positive gastric cancers show co-infection with EBV and that epigenomic analysis of EBV-infected gastric cancer cells revealed a hyper methylation of genomic DNA. In general, DNA hypermethlation results in the suppressed expression of the genes at the locus. Given this, we analyzed the epigenomic status of SHP1 gene and found that the gene locus is highly methylated in EBV-infected gastric cancer cells. We elucidated that the oncogenic activity of CagA is enhanced by epigenetic suppression of SHP1 in gastric cancer cells co-infected with H. pylori and EBV, resulting in enhanced tumorigenesis. These results provide a first demonstration that uncovered the phenomenon and molecular mechanisms in which bacteria and virus cooperatively promote cancer development.
Related paper: Saju et al., Nature Microbiology (2016)
7. Hit-and-run gastric carcinogenesis mediated by the CagA-driven "extrinsic BRCAness"
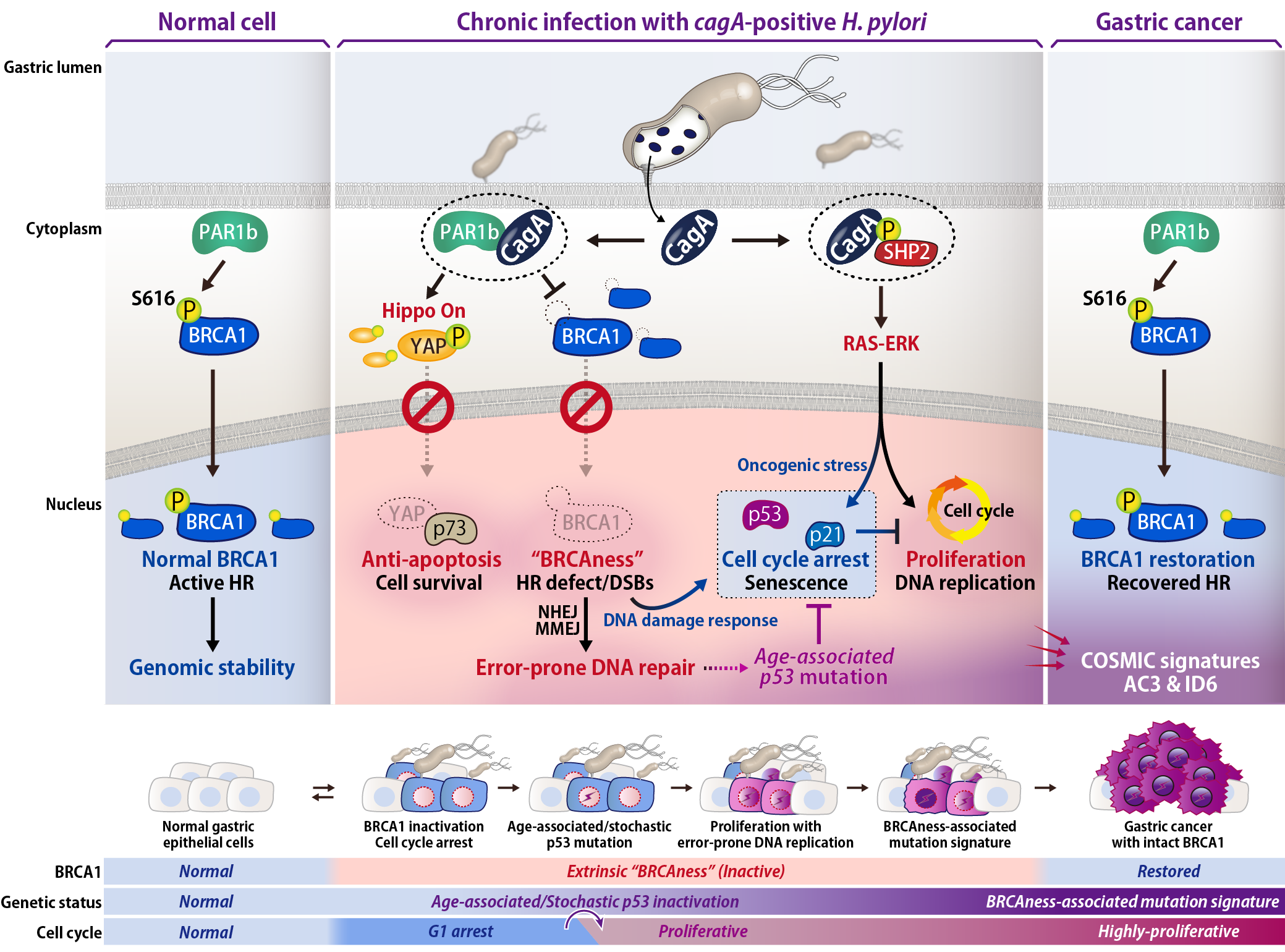
In H. pylori-associated gastric cancer, the carcinogenic bacteria no longer colonize at the cancer region, and therefore genetic and/or epigenetic alterations are required for maintenance of the transformed cells. This research demonstrated the molecular basis by which H. pylori CagA elicits an “extrinsic BRCAness” to promote accumulation of cancer-predisposing mutations in gastric epithelial cells.
First, we discovered that CagA induces DNA double strand breaks (DSBs) and cytoplasmic localization of tumor suppressor BRCA1, which primarily localizes to nucleus in normal cells. BRCA1 is deeply associated with DNA repair via homologous recombination and inactivated in several types of malignancies including breast and ovarian cancers. Further analysis revealed that PAR1b directly phosphorylated BRCA1 at Ser-616 near the NLS to control nuclear transport of BRCA1 and DNA repair, which were subverted by CagA-PAR1b interaction. We also found that CagA aberrantly activates Hippo signal to interfere with nuclear YAP-p73 interaction and DSBs-triggered apoptosis, contributing to survival of cells with genomic instability and thereby further promoting accumulation of mutations.
CagA promotes RAS-ERK signaling by activating SHP2, resulting in oncogenic stress, which induces p53/p21 to cease cell cycle. Hence we performed long-term and periodic expression of CagA in the gastric epithelial cells with p53 deficiency, which can be seen commonly with aging. As the result, the CagA-exposed p53 KO cells overcame the cell cycle arrest and underwent the BRCAness-associated genomic alteration (COSMIC signatures AC3 and ID6), which is characterized by impaired homologous recombination system. Intriguingly, intestinal-type gastric cancer cells also show the BRCAness-associated signatures. However, its mechanism was completely remained unclear since BRCA1 or its related genes rarely undergo mutations in gastric cancer. Based on our observations, we concluded that BRCAness-type genomic alteration found in gastric cancer arises from chronic inactivation of BRCA1 that is non-genetically caused by long-term exposure to the extrinsic effector protein CagA.
Collectively, this study uncovered the CagA-triggered molecular mechanism underlying the Hit-and-run process of cell transformation in which cancer-causing H. pylori promotes genetic alterations and eventually conducts infection-independent maintenance and progression of gastric cancer.
Related paper:Imai et al., Cell Host Microbe (2021)
8. Parafibromin, a novel substrate of SHP2
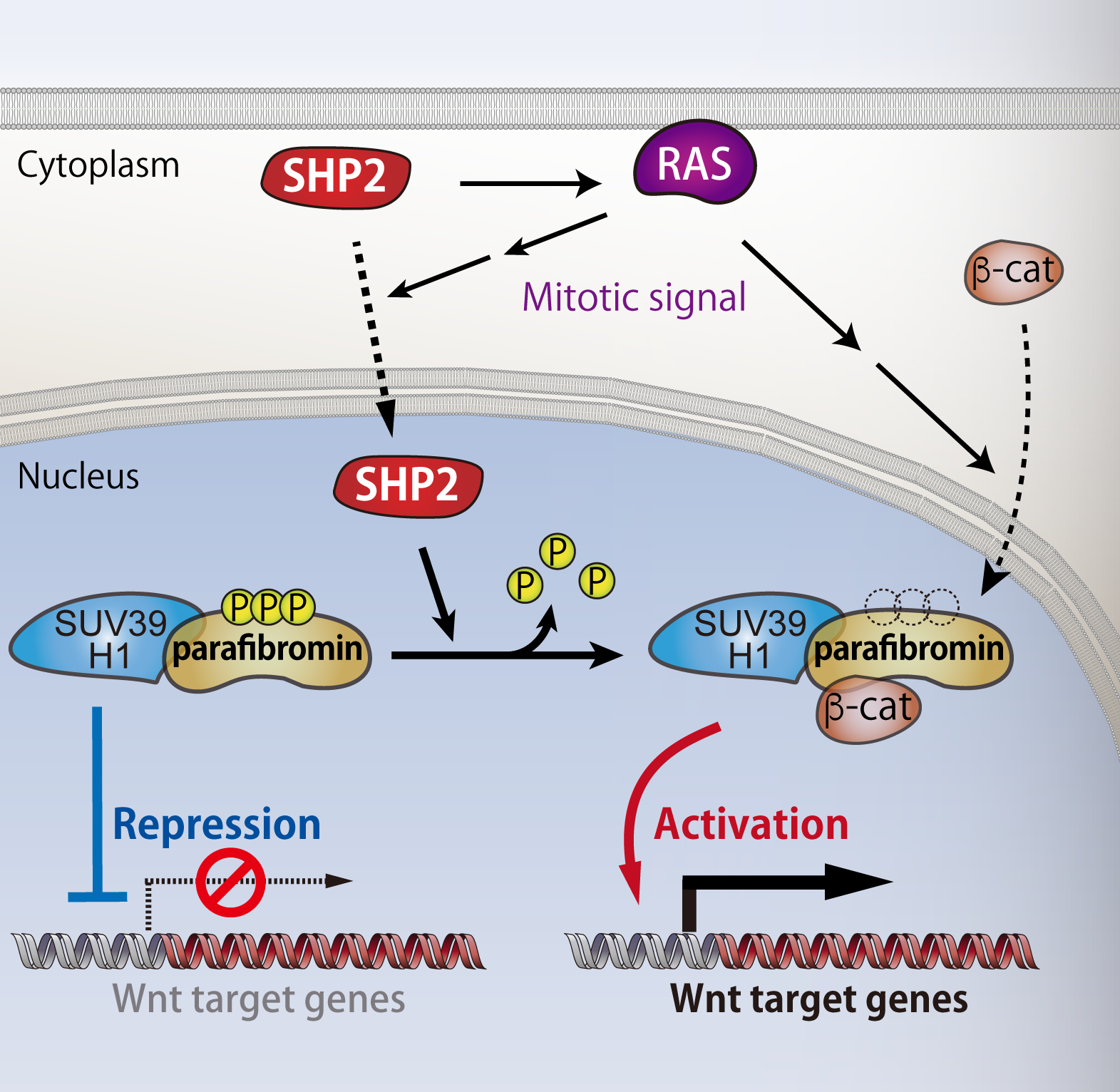
As SHP2 function has not been fully understood, we systematically explored novel substrates of SHP2 to investigate the unknown function of the phosphatase. As the result, we found that SHP2 dephosphorylates parafibromin, a key subunit of PAF complex that acts in transcriptional and post-transcriptional regulation in nucleus. Subcellular localization of SHP2 had been believed to be restricted in cytoplasm so far, however, this finding motivated us to investigate the SHP2 function in nucleus. A series of experiments revealed that upon activation of mitogenic RAS signalling, SHP2 re-localizes from cytoplasm to nucleus, where it dephosphorylates parafibromin. We also uncovered that the dephosphorylated parafibromin interacts with β-catenin and then activates Wnt signalling. In contrast to previous reports that showed tumor-suppressive role of parafibromin, our findings collectively indicates that parafibromin can act as a pro-oncogenic driver upon nuclear accumulation of SHP2 and subsequent dephosphorylation of parafibromin. Thus, SHP2 plays an important role in cell growth regulation in nucleus as well.
Related paper: Takahashi et al., Molecular Cell (2011)
9. Identification of an intracellular "logic board" implemented by Parafibromin
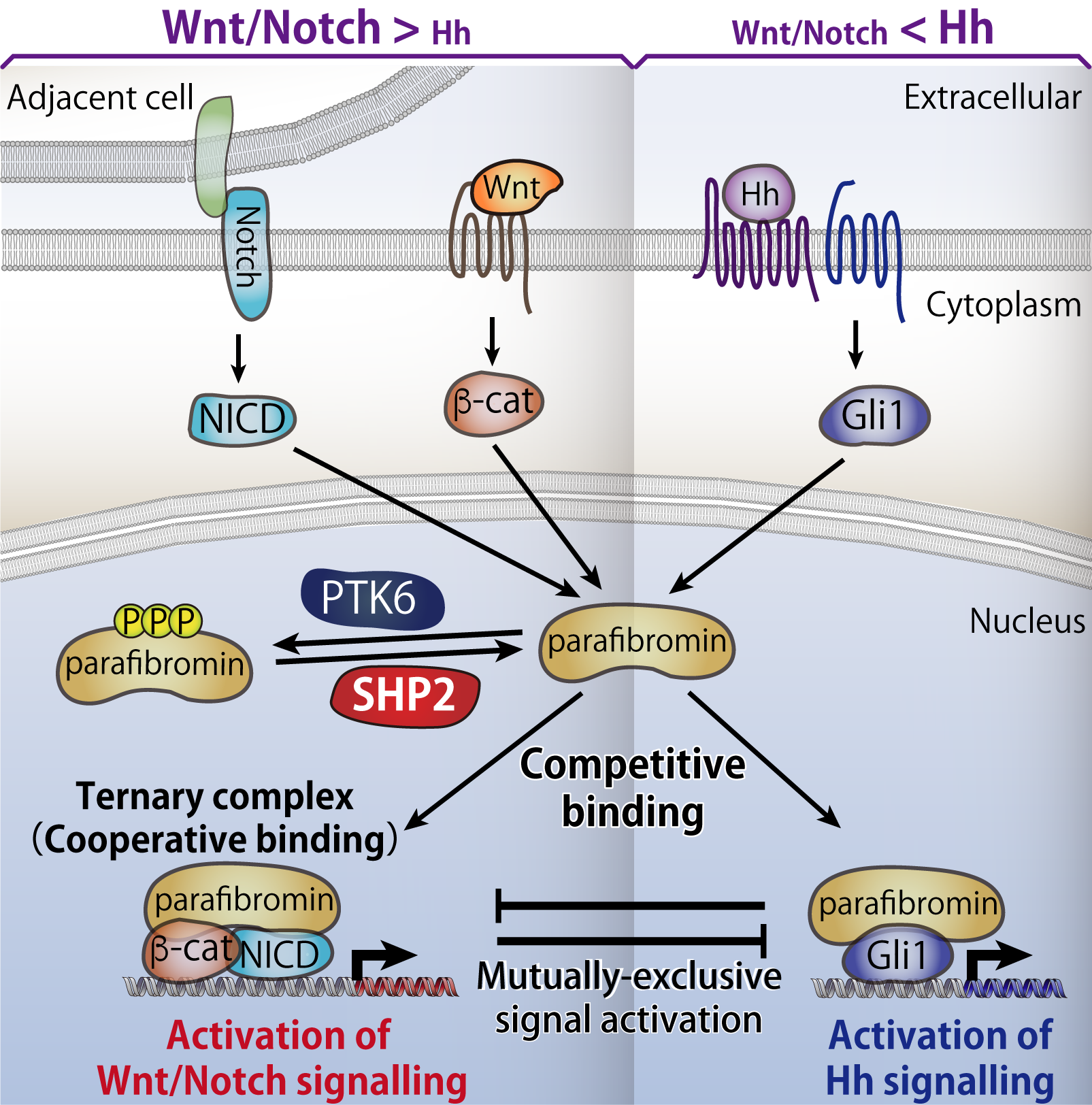
A variety of extracellular stimuli trigger a succession of signals transmitted along pathways within the cell, eventually giving rise to appropriate cellular responses such as cell proliferation and cell death. In multicellular organisms, morphogen signaling pathways, which help determine the structure and location of tissues and other materials, mediate communication between cells, thereby playing essential roles in embryonic development and maintaining internal stability, known as homeostasis, of adult tissues. However, very little is known about the mechanism through which a single cell perceives and integrates signals simultaneously activated in response to two or more distinct morphogens, the chemicals involved in cell development during tissue formation and differentiation, so as to generate appropriate transcriptional output and adequate cellular responses.
We discovered that parafibromin interacts with the transcriptional coactivators, proteins involved in transmitting genetic information, of three major morphogen signaling pathways (Wnt, Hedgehog, and Notch pathways)—either in a competitive or cooperative manner—and expresses genes appropriate to the cellular context. Thus, we concluded that parafibromin is a molecule that functions much like the logic board of a computer, integrating signal inputs transmitted by distinct devices and converting them into appropriate outputs.
Related paper: Kikuchi et al., Nature Communications (2016)
10. Subcellular localization and functions of SHP2 controlled by cell density
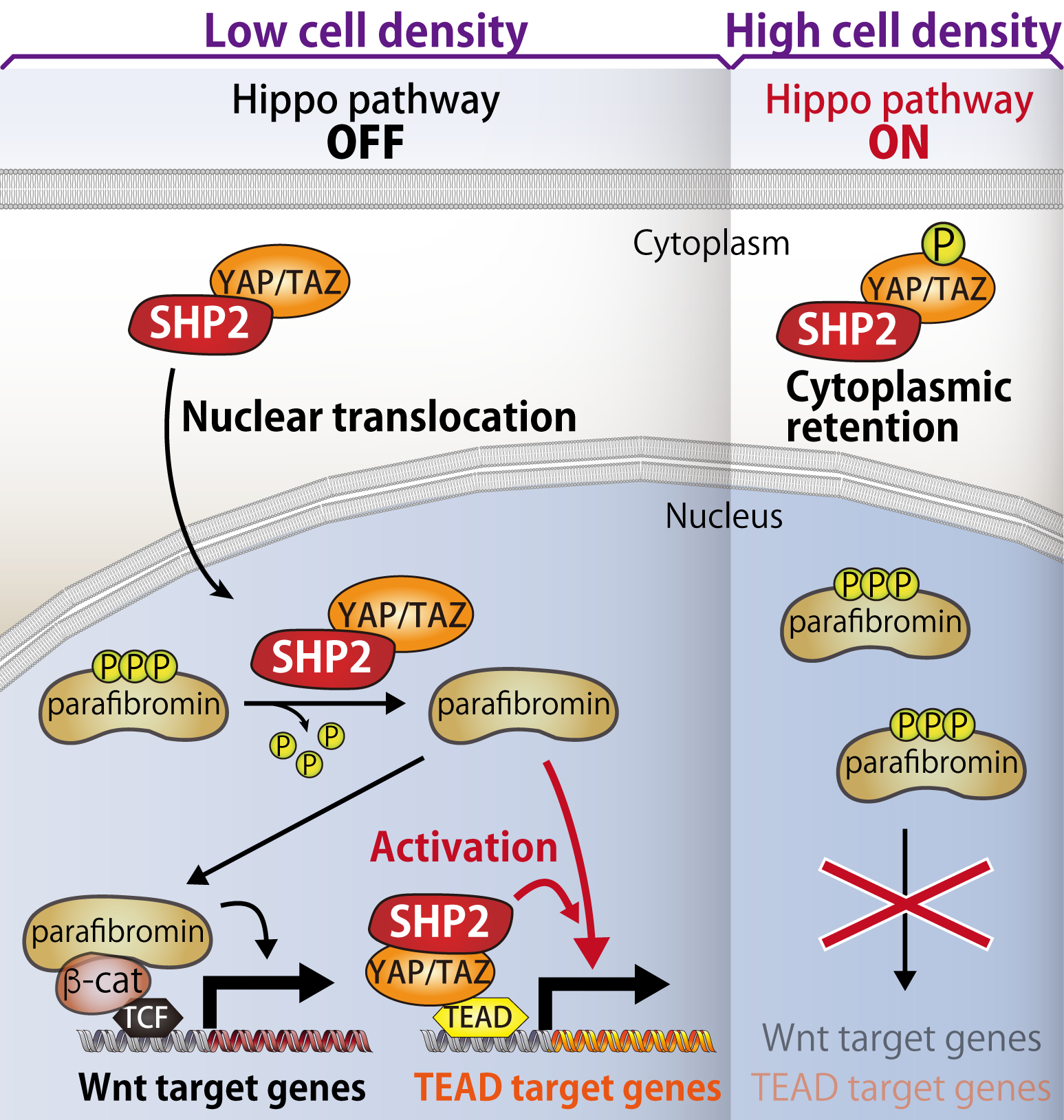
As nothing was reported about the molecular mechanism underlying nuclear accumulation of SHP2, we investigated how subcellular localization of SHP2 is controlled. We discovered that SHP2 binds to transcriptional co-activators YAP/TAZ, of which nuclear localization is regulated by Hippo pathway. Generally, proliferation of normal cells ceases when cells are highly contacting each other at a high cell density. This biological event, called "contact inhibition", is triggered by Hippo pathway, which is collapsed in many types of cancer.
During Hippo pathway is inactivated at the low cell density, SHP2 translocalizes to nucleous as a complex with YAP/TAZ and promotes transcription of Wnt- and TEAD-target genes. On the other hand, upon Hippo pathway is activated under the condition of higher cell density, YAP/TAZ are phosphorylated by Hippo pathway kinases and retained in cytoplasm, resulting in cytoplasmic retention of SHP2. Thus, nuclear function of SHP2 is suppressed during Hippo pathway is activated. Collectively, SHP2 is exploited by three distinct signalling pathways, proliferation-promoting RAS-MAPK and Wnt pathways and proliferation-suppressing Hippo pathway, to crosstalk with each other and manipulate the cell density-dependent cell growth.
Related paper: Tsutsumi et al., Developmental Cell (2013)
11. Collapse of Hippo pathway via hyaluronan degradation
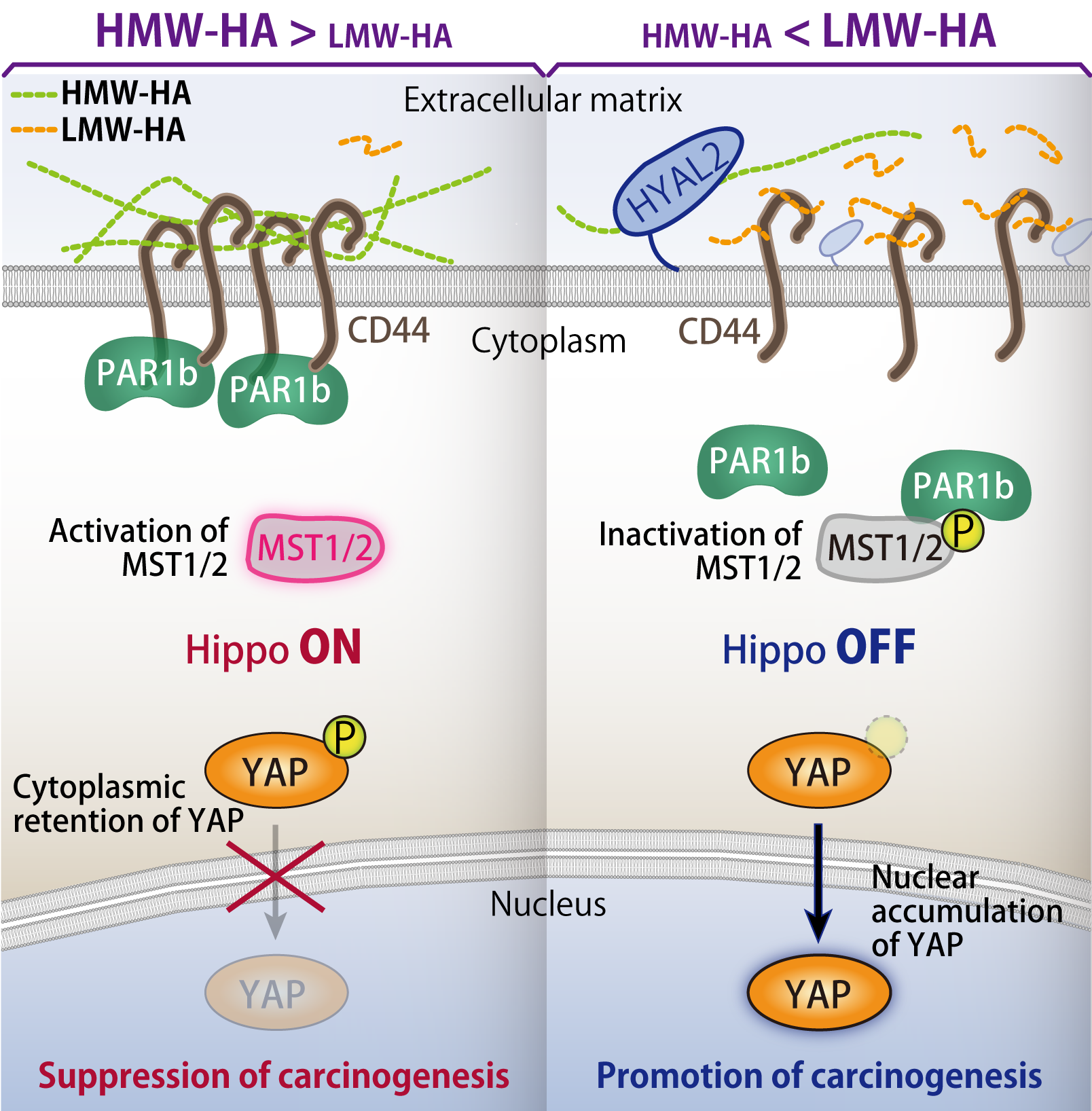
As described above, Hippo pathway acts as a tumor suppressive signalling cascade, by which cell growth is negatively regulated at the high cell density. However, the molecular mechanism how cells recognize extracellular signals to trigger Hippo pathway was unclear. Especially, the roles of extracellular matrices in Hippo pathway activation remained poorly understood.
We discovered that high-molecular-weight (HMW) hyaluronic acid (HA), an extracellular matrix molecule, induces cluster formation of HA-binding transmembrane protein CD44 to activate Hippo pathway. Whereas PAR1b can interact with MST1/2 kinases to inhibit Hippo pathway, clustering of CD44 leads to association of CD44-PAR1b complex and dissociation of PAR1b-MST1/2 complex, resulting in Hippo pathway activation. In contrast to HMW-HA, low-molecular-weight (LMW) HA inactivates Hippo pathway by promoting PAR1b-MST1/2 binding through a competitive inhibition of CD44 clustering.
HMW-HA is degraded to LMW-HA by HYAL2. We discovered that HYAL2 is frequently overexpressed in triple-negative breast cancer (TNBC), which is well known as highest grade of malignancy among breast cancer subtypes. Consistent with the elevated expression of HYAL2, we also found loss of HMW-HA and generation of LMW-HA as well as nuclear accumulation of YAP, indicating that Hippo pathway is collapsed in these HYAL2-overexpressing TNBC cells. Furtheremore, in vivo xenograft experiment demonstrated that HYAL2-overexpressing cells show inhibition of Hippo pathway via HMW-HA degradation, resulting in enhanced tumorigenic activity. Collectively, HA plays a crucial role in regulation of Hippo pathway in a molecular weight-dependent manner. This finding paves the way for development of new therapeutics by targeting HYAL2 or hyaluronan.
Related paper: Ooki et al., Developmental Cell (2019)
12. Distinct pro-oncogenic potentials of YAP1 regulated by alternative splicing
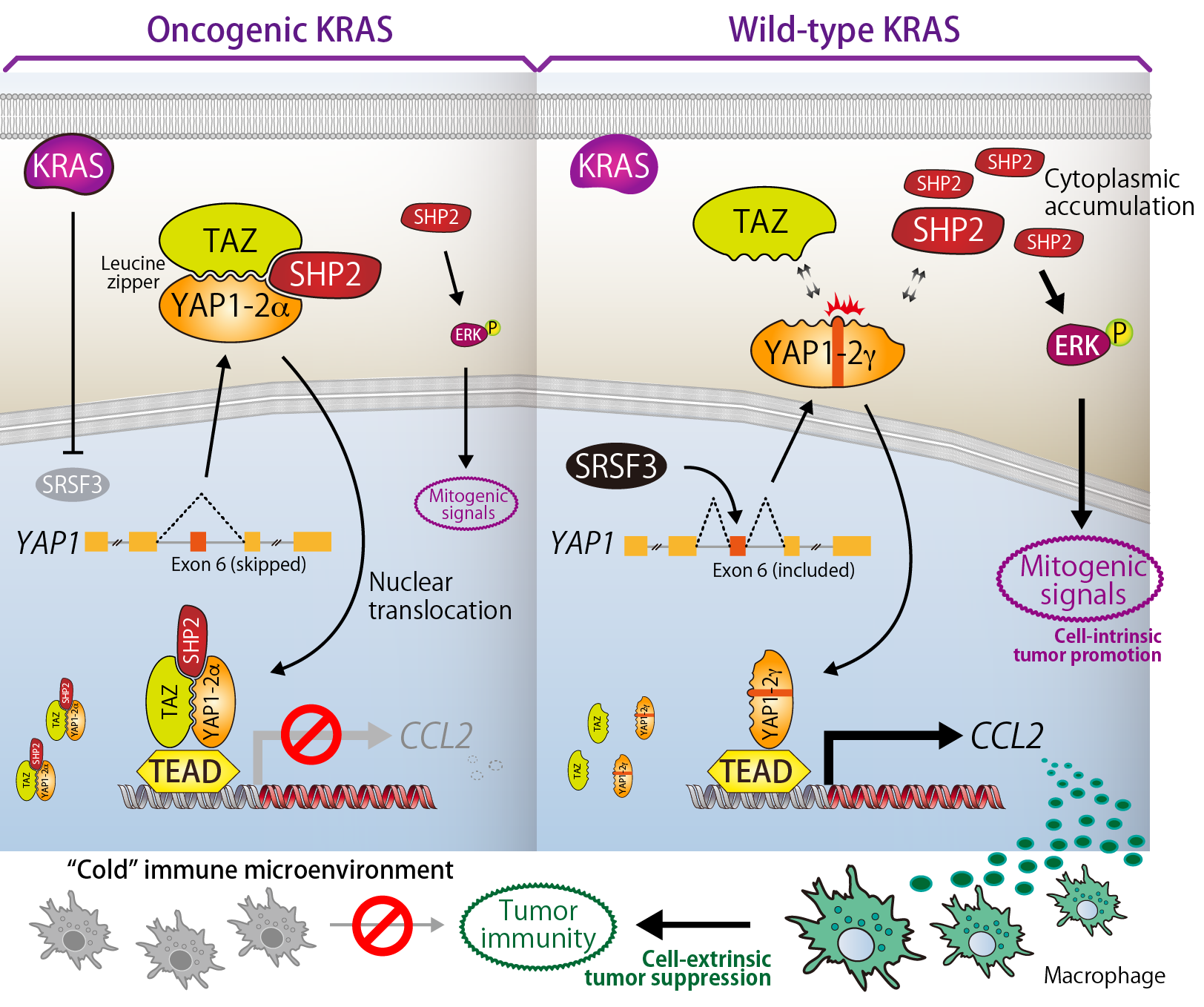
YAP1 gene (also simply called "YAP") comprises 9 exons and produces at least 8 YAP1 isoforms as splicing variants. This study found that functions of YAP1 are reversely controled by alternative splicing and demonstrated that the distinct actions of YAP1 isoforms cell-intrinsically and cell-extrinsically regulate the development/promotion of cancer.
TAZ and YAP1 possess the leucine zipper (LZ) motifs. The exon 6 of YAP1 encodes the γ-segment and disrupt the heptad repeats of leucines that are required for LZ formation between TAZ and YAP1. We investigated the functional and biological relevance of the exon 6 and found that YAP1-2α isoform, which does not contain the γ-segment, binds to TAZ via LZ structure to interact with SHP2. In contrast, YAP1-2γ containing the γ-segment interacts with neither TAZ nor SHP2. YAP1-2α translocalizes to the nucleus as a TAZ-YAP1-2α-SHP2 ternary complex and thereby suppresses cytoplasmic SHP2 activity that promotes the RAS-ERK signaling. Further analysis revealed that, in nucleus, the TAZ-YAP1-2α-SHP2 complex transrepresses CCL2, a monocyte/macrophage chemoattractant, resulting in an inactivation of tumor immunity. However, in YAP1-2γ-dominant cells, SHP2 is retained at the cytoplasm and nuclear translocated YAP1-2γ activates CCL2 expression to induce macrophage infiltration. Thus, alternative splicing of YAP1 regulates the cell-autonomous and non-autonomous prooncogenic activities.
We also identified SRSF3 as a splicing factor for YAP1 exon 6 and revealed that oncogenic KRAS induces the exon 6 skipping through down-regulating SRSF3 expression. Collectively, our research uncovered the significant biological relevance of YAP1-SHP2 interaction that plays a pivotal role in arrangement of "cold" immune microenvironment elicited by the cancer driver mutation.
Related paper: Ben et al., Journal of Biological Chemistry (2020)

 +81-3-3441-4173
+81-3-3441-4173  office@bikaken.or.jp
office@bikaken.or.jp